General
This section explains how to set up the Galaxy Docker container and use it to remove human traces from raw data and to submit reads to ENA. If you want to make use of an existing Galaxy instance that contains our tool, you can skip the first step. You can also find the content of this page on the Galaxy Training Network.
Walkthrough of reads submission to ENA using the Galaxy container
A screencast is available on our youtube channel that will guide you through all the steps discussed below:
1. Setting up the Galaxy container
- Open the terminal
- Make sure docker is installed and available on your path. To check this, simply type
docker run hello-worldin the Terminal or and press enter. If Docker is installed it will give some usage information back. For more information on how to install docker please visit this website. - Run following command in the terminal to run the container:
-
Linux/Windows:
Raw read submission + assembly workflows:
docker run -p "8080:80" --privileged quay.io/galaxy/covid-19-uploadRaw read submission only:
docker run -p "8080:80" --privileged quay.io/galaxy/ena-upload -
MacOS:
Raw read submission only:
docker run -p "8080:80" quay.io/galaxy/ena-upload:hg38
-
- Wait a minute or two until the container is started
- Visit http://localhost:8080/ to access your local instance of Galaxy
More information about the container can be found in the github repository.
2. Obtain and load ENA Webin submission credentials
An ENA Webin account is required to upload data to the ENA. If you plan to offer this tool as a service to multiple users, then a brokering account is more suitable. To change your Webin account into a broker account, please contact ENA.
You can load your ENA credentials in Galaxy:
- Login as admin using
adminas username andpasswordas password, this will give you full access to the galaxy instance. - Go to User > Preferences in the top navigation
- Click on Manage Information
- Fill in the
Your ENA Webin account details - Click Save
3. Upload data to Galaxy
This tool is used to submit raw reads to the ENA. Genome or transcriptome assemblies can be submitted to ENA using their website. The submission tool currently accepts fastq.gz (SE and PE) file format.
Raw data can be uploaded using Galaxy’s upload tool found at the top right of the Tools panel (Fig. 1a) or at Upload File from your computer under Upload Data tool group (Fig. 1b).
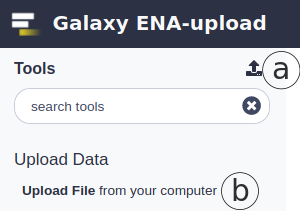
Figure 1. Data upload locations in Galaxy
There are different options for uploading data files:
- Local files can be uploaded by drag-and-dropping to ‘Drop files here’ (Fig. 2a) area or by selecting ‘Choose local files’(Fig. 2b) and browsing to the local files
- ‘Paste/Fetch data’ (Fig 2c) allows fetching remote files from the web by entering a URL or pasting the data itself
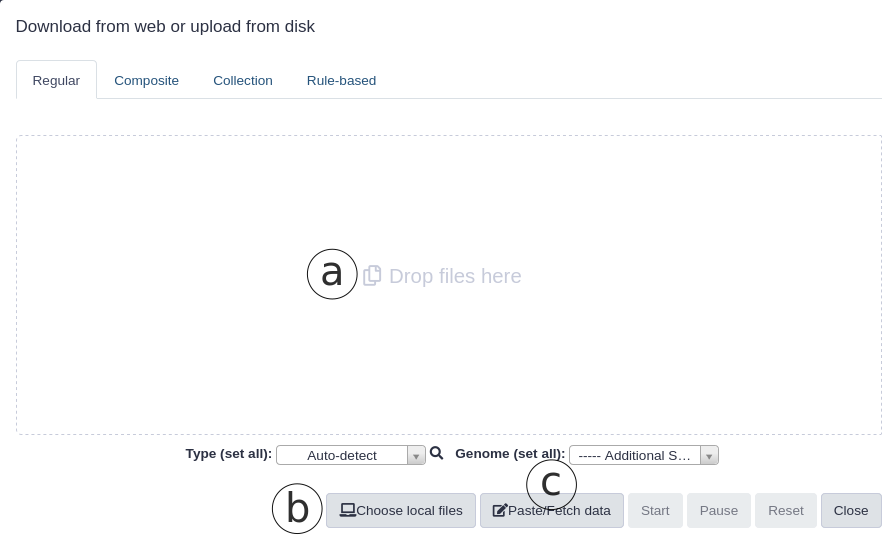
Figure 2. Data upload options in Galaxy
When the local files are selected or a URL is given, click “start” to start the uploading to Galaxy. More information on data upload to Galaxy can be found in Galaxy support.
Your data should appear in green on the right history panel (Fig. 3). You can rename, tag, preview edit or delete data objects from here.
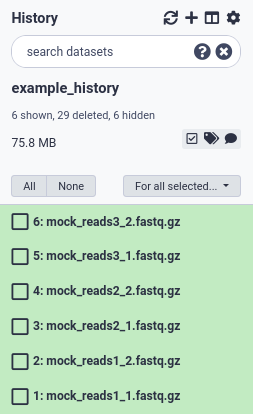
Figure 3. Files that are uploaded will show up in the history panel.
The files need to be in fastq.gz or fastq.bz2 compressed format. Galaxy will try to guess the datatype during upload. If it fails to do so correctly, you can Edit the dataset’s attibutes and manually change the datatype to fastq.gz
4. Filter human reads out of the raw reads
In order to comply with Europe’s General Data Protection Regulation (GDPR), traces of human genetic information must be removed from the raw data before submitting it to ENA. A tool is included that filters out reads that map to the human genome using Metagen-FastQC.
Select the filtering tool from the Tools panel on the left.
- Select human h38 reference genome (Fig. 4a).
- Choose single or paired-end (Fig. 4b)
- Select the files to clean (uploaded on the previous step, Fig. 4c). These can be single or multiple files or a collection of files.
- Click on Execute (Fig. 4d)
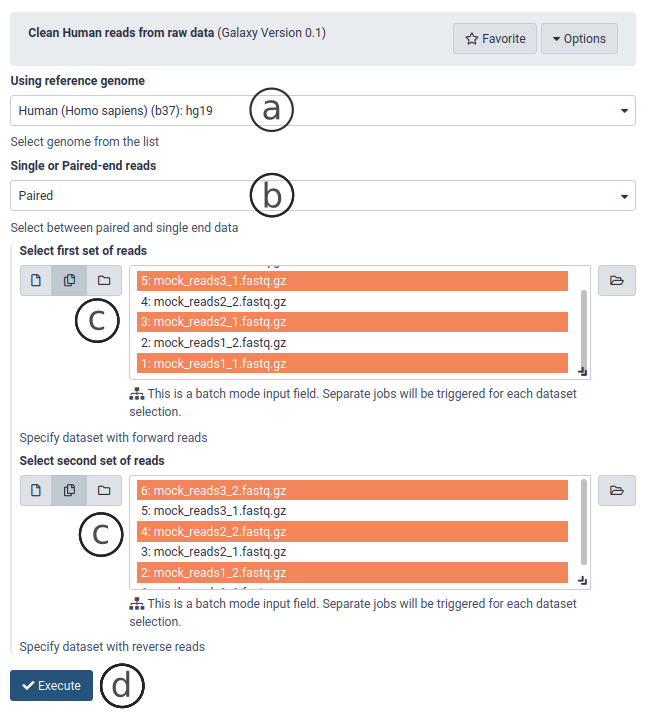
Figure 4. The interface of the read cleaning tool.
The tool will now process the raw reads to remove reads that map to the human genome. This can take a while. The resulting filtered data files are found on the right panel.
Optional: create a collection
Galaxy collections help you organize your data and minimize your history clutter. If you plan to use the Galaxy COVID-19 genome analysis workflows you must organize your reads data into collections. You can make collections for single end or paired end datasets.
To make a collection of paired-end datasets, first select all PE datasets and select Build List of Dataset Pairs for all selected (Fig. 5). The names of the PE read files should be in the format ‘pair_name’ + ‘_1.fastq.gz’ for the forward reads and ‘pair_name’ + ‘_2.fastq.gz’ for the reverse reads. Galaxy will recognize the _1/_2 suffix for PE reads (Fig. 6a). This is also the standard for PE file naming in ENA. If a different schema was used, it is best to change the filenames to the _1.fastq.gz/_2.fastq.gz naming schema. Make sure the names in the metadata are identical (see below) to the filenames. The datasets are paired by name (Fig. 6b), and you can name the collection (Fig. 6c). Finally, click on Create List (Fig. 6d).
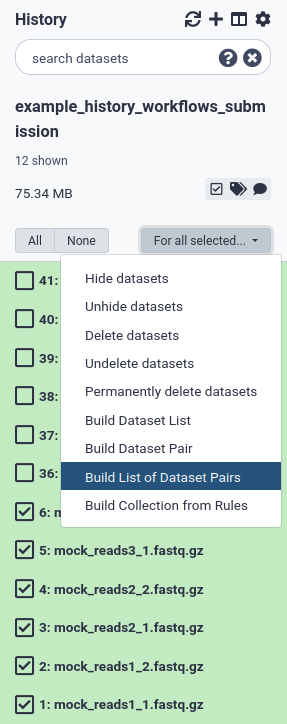
Figure 5 Select datasets for paired-end collection (list of dataset pairs) .
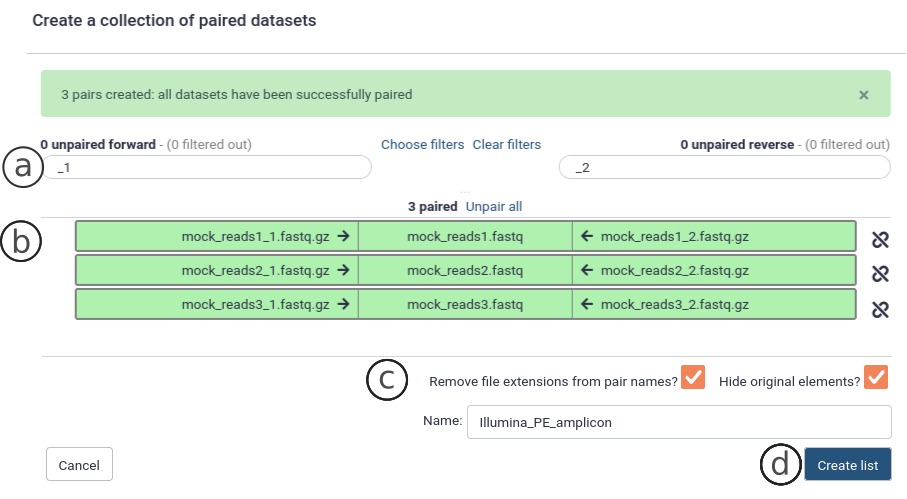
Figure 6 Create a collection of paired datasets .
5. Upload metadata and submit to ENA
The ENA Upload tool under Submission tools is used to generate the metadata in the right format, associate it with the sequence data files and submit everything to ENA.
Execution mode
By default the tool is set to add new data, which will add new metadata objects and data to ENA. By changing the Action to execute dropbox to modify, one can also modify his existing entries on ENA.
Testing options
It is advisable to first test your submissions using the Webin test service where changes are not permanent and are erased every 24 hours. Do this by selecting ‘Yes’ on ‘Submit to test ENA server?’. There is also the possibility to run the client side metadata validation without submitting to the ENA servers by selecting ‘Yes’ on ‘Print the tables but do not submit the datasets’.
Metadata upload
Submission to ENA requires accompanying metadata that complies with the ENA metadata model.
The tool offers three ways of entering metadata for submission:
- Using the metadata spreadsheet template (default, Fig. 7a)
- Interactively generating the metadata structure (Fig. 7b)
- Using 4 tsv tables (legacy, Fig. 7c)
All three allow you to make a submission to either the test or production server of ENA. All metadata fields must be completed for the submission to go through. The submission tool will validate the metadata before submission.
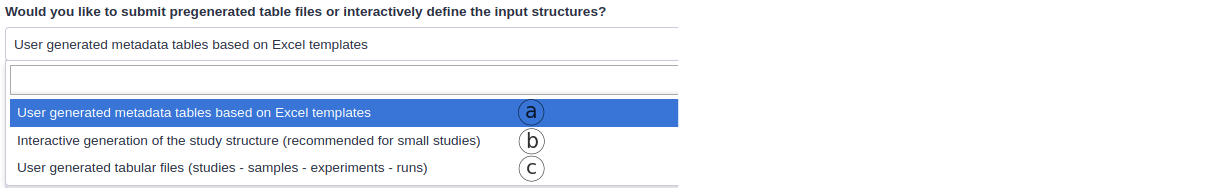
Figure 7. Three ways of submitting the metadata through the ENA-upload tool.
The spreadsheet, interface and tsv tables are organized following the ENA model:
- A study (project) groups together data submitted to the archive and controls its release date. A study accession is typically used when citing data submitted to ENA.
- A sample contains information about the sequenced source material. Samples are associated with checklists, which define the fields used to annotate the samples. Samples are always associated with a taxon.
- An experiment contains information about a sequencing experiment including library and instrument details.
- A run is part of an experiment and refers to data files containing sequence reads.
Use the _alias field on each sheet to interlink the experiments, studies, runs, samples and files with each other. The template can be downloaded, completed and uploaded using the Galaxy upload tool.
Spreadsheet template upload
For submission of a large number of files, we recommend to use the Excel template to upload the metadata. The spreadsheet contains one worksheet each for study, sample, experiment and run metadata. A template repository is in place that supports a spreadsheet with controlled vocabulary build in for every checklist on ENA.
Interactive metadata upload
For a small number of submissions, metadata is best entered interactively in Galaxy using the submission tool. Metadata fields are nested according to ENA metadata model described above.
Tabular (tsv) metadata tables (Legacy)
Four tsv files can be uploaded, one each for study, sample, experiment and run metadata. Example tsv files can be found Export the completed tables to tsv files and upload them using Galaxy upload tool.
Complete the metadata template (spreadsheet and tsv tables) in your computer. All fields of the template must be complete. Here you can find an example of part of the metadata associated with an actual submission to ENA (accession number PRJEB40711). Upload the metadata file to Galaxy, and select it on ‘File based on templates here:’. Finally, select the human-filtered data files associated with the metadata, fill in the Affiliation center and click on ‘Execute’.
Select the correct checklist
You can specify the ENA sample checklist using the dropdown. The supported checklists are described on the ENA website. If you want to submit SARS-Cov-2 raw reads, you have to select the ENA virus pathogen reporting standard checklist (ERC000033) checklist in the dropdown menu as seen in Figure 8.
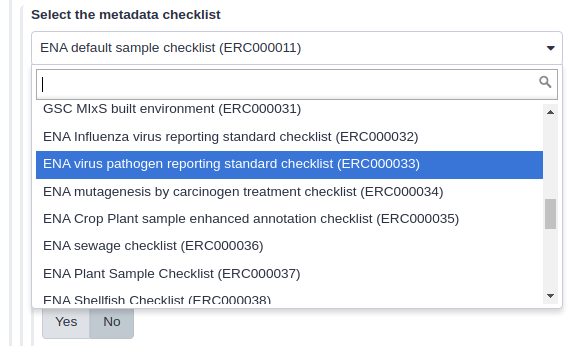
Figure 8. Select the correct ENA metadata checklist
Select data and submit
Next, select the human-filtered data files associated with the metadata and select the correct metadata file for each section. This tool accepts individual datasets, mixed collections or paired-end collections.You can make collections for single end or paired end datasets.
Finally, fill in the Affiliation center and click on ‘Execute’. The output of a successful submission in Galaxy are four metadata tables (Fig. 8a) and a submission ‘receipt’ (Fig. 8b).
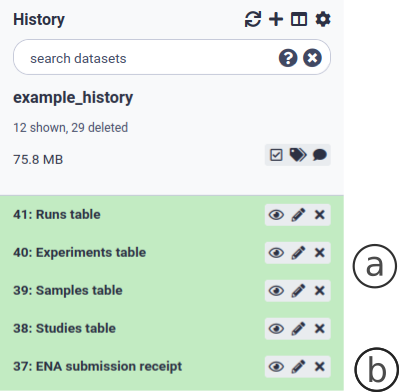
Figure 9. Output of a successful reads submission to ENA. The submission receipt contains metadata used for consensus sequence submission.
6. Check for a valid submission
Visit Webin online to check on your submissions or dev Webin to check on test submissions. If everything looks fine, publish the data by changing the “Release Date” of the study to a day later than the current day. It can take several days for ENA to index the data and to let it appear in a correct manner. Covid-19 data will also be indexed by the COVID-19 Data Portal