General
This second part of the documentation explains how to run the COVID-19 analysis workflows to generate a consensus sequence and how to submit it to ENA.
About Galaxy workflows
A workflow is a series of tools and dataset actions that run in sequence as a batch operation. Galaxy workflows can be annotated, viewed, shared, published, and imported. They ensure reproducibility of analyses. Maier et al. (2001) have produced Galaxy workflows for the analysis of Illumina WGS and amplicon as well as ONT amplicon SARS-CoV-2 data. Workflows are found under the workflow tab in the top navigation and at the bottom of the tools menu.
7. Run the variation analysis workflow
There are currently four workflows for variation analysis that cater for different libraries/platform combinations:
- Illumina whole genome sequencing (WGS) paired-end (PE) or single-end (SE)
- Illumina amplicon (ARCTIC protocol) paired-end
- Oxford Nanopore amplicon (ARCTIC protocol)
If you have not already done so, upload your reads data as explained in section 3. Go to the workflow section and select the variation analysis workflow suited for your data by clicking on . This brings up the workflow menu. The ARCTIC variation analysis workflow requires:
- Your data as a collection in fastq.gz format of all samples to be analyzed (Fig. 9a).
- The SARS-CoV-2 reference FASTA sequence, included in the container (MN908947.3.fasta - (Fig. 9b))
- A BED file describing the binding sites of all primers used to generate the tiled amplicons (Fig. 9c). We include in the container the v3 primer scheme BED file (other versions can be obtained from the ARTIC project repo)
- An amplicon info file. This tabular file should consist of one line per amplicon, which should list all primers involved in the generation of this amplicon (Fig. 9d). We include in the container the amplicon info for the v3 primer scheme (other versions can be obtained from the ARTIC project repo).
These necesary files are also found in useGalaxy.eu. The workflows to analyze whole genome sequencing (RNA-Seq) library-derived sequences do not require the last two files. The outputs of the variation analysis workflow are a alignment file (bam) collection (Fig. 10b), a report on the alignments (html - Fig. 10d) and a Variant Call File (vcf) collection (Fig. 10a). Both are needed to construct the consensus sequence.
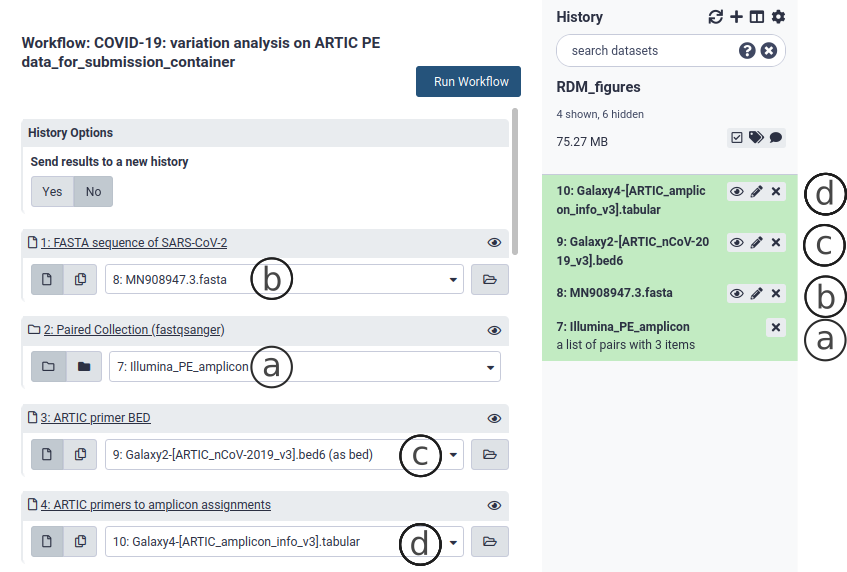
Figure 9. The variation analysis workflow (ARCTIC Illumina PE).
8. Run the consensus construction workflow
Go to the workflow section and select the Consensus Construction workflow. This workflow takes as input a Variant Call File (vcf) collection (Fig. 8a), an alignment file (bam) collection (Fig. 10b) and SARS-CoV-2 reference genome (Fig. 10c). The first two are produced by any of the variation analysis workflows described above in section 7 and are found in your history. The SARS-CoV-2 reference genome is included with the container and will also be found in your history. Select the appropriate collections and files and Run Workflow. This workflow outputs a consensus sequence collection as well as one multisample fasta with all the consensus sequences.
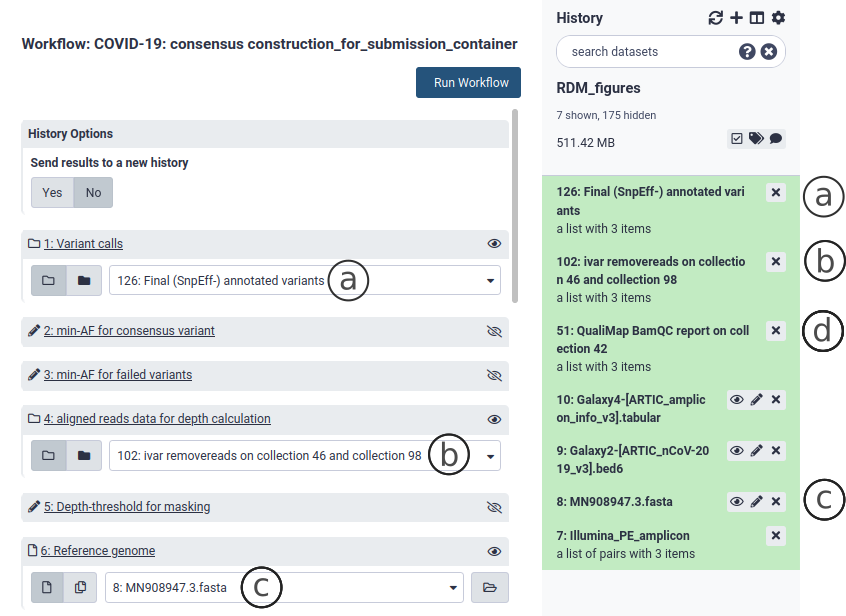
Figure 10. The consensus construction workflow.
9. Submit consensus sequence to ENA
Open the consensus sequence submission tool This tool requires a fasta file (single submission) or a multi-fasta file (multiple submissions) as well as the associated metadata.
Metadata
In most cases, the Assembly type will be Isolate and Molecule type will be either genomic RNA or viral cRNA, depending on your library preparation strategy.
If the consensus was generated using the Galaxy workflows described in section 7 and 8, then the Assembly program and Coverage metadata can be found in the alignment report (html) file Fig. 8d.
You can choose to input the rest of the metadata interactively or if the reads were submitted using the Galaxy reads submission tool, a metadata receipt was produced that contains most of the metadata needed to submit consensus sequences to ENA, including Study and Sample accession numbers. This metadata is linked to the consensus sequences by the original reads filename and can be parsed to extract the metadata associated with the reads/consensus.
Visit Webin online to check on your submissions or dev Webin to check on test submissions as explained in section 6.
10. Stopping and deleting the container (optional)
The container can be stopped by simply closing the terminal. In this case the container will still be there. If your submission was successful and you don’t need the instance anymore, you can delete the container and its corresponding image using following steps:
-
Check the name of your container you want to delete
docker container list -a -
Delete the container using its name (seen in the NAMES column)
docker container rm NAME -
Delete the image (when you don’t want to use the galaxy instance in the foreseeable future)\
-
Linux/Windows:
Raw read submission + assembly workflows:
docker rmi quay.io/galaxy/covid-19-uploadRaw read submission only:
docker rmi quay.io/galaxy/ena-upload -
MacOS:
Raw read submission only:
docker rmi quay.io/galaxy/ena-upload:hg38
-